Irisappan Sarath Chandran1,
Pichandy Muthu Prasanna2 ![]()
For correspondence:- Pichandy Prasanna Email: muthuprasanna1000@rediffmail.com Tel:+919908947749
Received: 5 March 2016 Accepted: 2 October 2016 Published: 28 November 2016
Citation: Chandran IS, Prasanna PM. Formulation and evaluation of analgesic activity of polysorbate 80-coated loperamide liposomes in mice. Trop J Pharm Res 2016; 15(11):2297-2302 doi: 10.4314/tjpr.v15i11.1
© 2016 The authors.
This is an Open Access article that uses a funding model which does not charge readers or their institutions for access and distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0) and the Budapest Open Access Initiative (http://www.budapestopenaccessinitiative.org/read), which permit unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited..
Purpose: To deliver loperamide (Lp) into mice brain using polysorbate 80 (PS80)-coated liposomes that inhibits P-glycoprotein (P-gp) efflux.
Method: Lp loaded liposomes were prepared by reverse phase evaporation technique using lecithin (Lec) and cholesterol (Ch). The efficacy of PS80-coated Lp liposomes (PLs) in mice was evaluated using central analgesic models (Eddy’s hot plate method and tail immersion test) and peripheral analgesic model (acetic acid-induced writhing).
Results: PLs showed maximum possible response (MPR) of 58.33 % at 60 min in Eddy’s hot plate study. In the tail immersion test, PLs showed MPR of 67.64 and 69.24 % at 60 and 90 min, respectively, relative to control group. This confirms the potential of PLs to deliver Lp to the brain by inhibiting P-gp efflux. Dose response study using tail flick method confirmed the minimum Lp dose (25 µg/kg, i.v) required to achieve central analgesic activity using PLs.
Conclusion: PS80-coated Lp loaded liposomes (PLs) possess a good potential to inhibit P-gp efflux of Lp from brain, and also exhibit both central and peripheral analgesic activity.
Introduction
Blood brain barrier (BBB) and cerebrospinal fluid barrier (CSFB) control the inflow of drug molecules into the brain. Lack of endothelial fenestrations and the presence of endothelial tight junctions enable BBB to restrict the drug transport into the central nervous system [1]. The BBB removes or effluxes most of the macromolecules unless specific carriers, transporters or receptors are present in the endothelial cells. Essential molecules such as glucose, amino acids and nucleosides are transported by specific proteins in the luminal surface of the endothelial cells and the transport of essential proteins and peptides to the brain are mediated by transcytosis [2,3].
P-glycoprotein (P-gp) is a potent peptide in BBB which prevents any untoward drug or molecule entry in to the brain by its efflux nature [4]. Loperamide (Lp) is a morphine like opioid receptor agonist. Due to P-gp mediated efflux, Lp cannot enter into the brain thereby preventing its central analgesic activity. It acts on gastrointestinal µ and δ opiate receptors and reduces gastrointestinal motility [6]. Earlier research reported that Lp delivered by human serum albumin nanoparticle with adsorbed monoclonal antibody produces anti-nociceptive activity in tail flick test [7]. P-gp inhibitors such as indole alkaloids, quercitin, surfactants were also studied to assess their ability to facilitate BBB penetration [8].
Kreuter et al reported that the surfactant Polysorbate 80 (PS80) enabled the drug dalargin to reach brain by inhibiting P-gp efflux and produce anti-nociceptive activity [9]. PS80 adsorbs apolipoprotein- E globules over the drug to deliver it from the blood. PS80 mimics the carrier as LDL particle and LDL receptor recognize it as its ligand, favoring its entry in to brain by endocytosis [10]. Drugs such as tacrine, doxorubicin, hexapeptide and tubocurarine were also targeted using PS80 surfactant [12].
Thus PS80 adsorption or coating could help us in delivering loperamide to the brain. Liposome offers a great advantage of entrapping both hydrophilic and hydrophobic drug and its lipid constituents further promotes endocytosis through BBB. Its nano-sized dimension synergizes its brain targeting potential. Surface modification, antibody conjugation, stealthing etc. of liposomes makes it a potential carrier for brain targeting molecules [15].
Thus, in the present study, an attempt has been made to prepare PS80-coated Lp liposomes and evaluate its central analgesic activity.
Methods
Materials
Polysorbate-80 was obtained from Sigma Aldrich and loperamide was received as a gift sample from Alembic Pharmaceutical, Vadodara, Gujarat. Lecithin, cholesterol and vitamin E were of analytical grade.
Formulation of PS80 coated and uncoated Lp liposomes
Lp loaded liposomes were prepared by “Reverse Phase Evaporation Technique” using lecithin (Lec) and cholesterol (Ch) with modification [16]. The lipids, Lec and Ch, (in 9:1 ratio) were dissolved in diethyl ether. Drug solutions of Lp (5 mL, 2 mg/mL) were prepared by dissolving Lp in chloroform along with 0.5 mL of vitamin E (0.6 mole %) to prevent lipid oxidation. Using the homogenizer (Tenbroeck tissue grinder country; operated at 5000 rpm, for 20 min, at 50 °C) the solution was emulsified to form liposomal suspension with semi-solid gel like consistency until the chloroform evaporates. Any trace chloroform was evaporated using a vacuum evaporator (BUCHI EL 131 Rotavapor, Germany) under reduced pressure (260 - 400 mmHg) at 60 °C. The lipid gel was then collapsed and vortexed to attain fluid like consistency with addition of 10 mL phosphate buffer solution (PBS) and sonicated using a micro-tip probe sonicator(30 min; 40 % frequency), (Vibra-Cell, Sonics and Materials, Inc., Danbury, CT, USA) forming multi-lamellar vesicles (MLV) in the liposomal suspension. PS80 coating was done by adding PS80 (2 %) drop wise in the MLV liposomal suspension with continuous magnetic stirring at 9000rpm; for 45 min followed by centrifugation (75,000 g for 20 min). The supernatant free Lp, excess PS80 and PEG were discarded. Trace oxygen was removed by flushing nitrogen gas and the PS80 coated MLVs (PLs) were lyophilized and stored at 4 °C for further study. Uncoated MLVs of loperamide (UL) were prepared as above but without the PS80. Blank PLs (BPLs) were prepared using the same protocol described above in the absence of Lp.
Experimental animals
Healthy Swiss albino mice weighing 20-25 g of either sex were maintained under controlled conditions of temperature at 23 ± 2 °C, humidity 55 - 60 % and a 12h light–dark cycle. They were housed in sanitized polypropylene cages containing sterile paddy husk as bedding. They had free access to standard feed (standard commercial pellet diet) and water ad libitum. Experimental protocols were reviewed and approved by the Institutional Animal Ethics Committee – Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Prist University (Registration No. 292/CPCSEA/PHARMCEUT-11/06) and were performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals(US) [29].
Evaluation of analgesic activity
Eddy’s hot plate method
Eddy’s hot plate method was to evaluate thermally-induced pain in mice. In this method mice were divided into eight groups (each group n=10). Mice in control group were injected with phosphate buffer solution (2 mL/kg,i.v). Mice in *Lp group were injected with 10 μL of intrathecal Lp formulation (3 μg/mL) [17] through intrathecal catheter and flushing with physiological saline (0.9 %) [18]. Mice in $Lp group were injected with intravenous Lp formulation (0.03 mg/kg, i.v) [19] through tail vein. Mice in PLs group were injected with PS80 coated Lp Liposomes (0.03 mg/kg, i.v). Mice in UL group were injected with uncoated Lp liposomes (0.03 mg/kg, i.v). Mice in PLp and BPLs groups were injected with Lp dispersed in 2 % PS80 solution and PS80 coated liposomes without Lp at the dose of 0.03 mg/kg, i.v and 2 mL/kg, i.v respectively. NPLs group received Naltrexone HCl (0.1 mg/kg, i.v) along with PS80 coated Lp liposomes (0.03 mg/kg, i.v).
Eddy’s hot plate (Columbus Instruments, Columbus, OH) maintained at 52 °C + 0.5 was used to evaluate response to thermal-induced pain. The “Maximum Possible Reaction Time” (MPR) of each mouse is the measure of hind paw withdrawal from the Eddy’s hot plate (thermal withdrawal latency) and is the time interval between placement on the hot-plate and the first jump or lick of a hind limb. Animals with average baseline latency less than 25 s were used in this study. A cut-off latency of 30 s was followed to avoid tissue damage or thermal hyperalgesia [20]. The formulations and PBS were administered 30 min before the evaluation of thermal-induced pain and the MPR was recorded at 30, 60, 90 and 120 min.
The degree of analgesic activity was expressed as percentage of maximal possible response (MPR in %), and was calculated as follows:
MPR(%) = ([Test-Control])/Test X 100 ……. (1)
Tail immersion test
In this method the mice were divided into eight groups (each group n = 10) and were prepared as explained above for the Eddy’s hot plate technique. The lower 5 cm portion of each tail was immersed in a beaker of water maintained at 58 ± 0.5 °C. The cut off time of 20 s was maintained to avoid damage to the tail. The formulations and PBS were administered 30 min before the evaluation of tail immersion test and the MPR was recorded at 30, 60, 90 and 120 min [9]. The degree of analgesic activity was expressed as percentage maximal possible response (MPR %), and was calculated using equation 1.
Acetic acid-induced writhing test
In this method the mice were divided into eight groups (each group n =10) and were prepared as explained above for the Eddy’s hot plate technique.
The formulations and PBS were administered 30 min before the acetic acid administration and the total number of writhing following intraperitoneal administration of acetic acid solution (1 % v/v in normal saline, 10 mL/kg) was recorded over a period of 10 min, starting 5 min after acetic acid injection [22]. The percentage protection was calculated according to the equation 1.
Dose-response studies
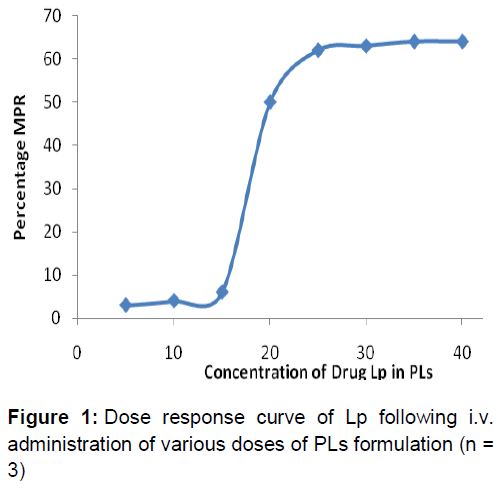
Based on the result of in vivo studies, PLps was selected due to its effective central analgesic activity for determining the minimum Lp dose required for maximum MPR% response. Lp concentrations ranging from 5 µg/kg (6 times less from the IV Lp dose) to 40 µg/kg were selected. Lp equivalent to 5, 10, 15, 20, 25, 30, 35 and 40 µg/kg loaded in PLs were administered to animals by IV and observed for its analgesic activity by tail immersion test [23]. The response was plotted as MPR% as described above and curve was constructed ().
Statistical analysis
The data for analgesic activity was expressed as mean ± standard deviation (SD). Statistical analysis was carried out using one-way ANOVA followed by Tukey’s t-test. Differences were considered significant at p ≤ 0.05.
Results
Analgesic activity
The MPR% of various formulations and PBS are shown in . The response time for all the observations for both Eddy’s hot plate and tail immersion test was 2 h. A base line response was recorded for PBS treated mice in control group. In Eddy’s hot plate method *Lp showed maximum MPR% at 1 h (60.22 %) followed by PLs (58.33 %).
Tail immersion test also reflected the same response with higher MPR% at 1 h for *Lp (73.21 %) followed by PLs (67.64 %). Intrathecal delivery of Lp (*Lp), delivered the drug directly to brain eliciting analgesic effect of Lp over this noxious thermal stimulus. PLs also exhibited 67.64 % and 69.24 % MPR at 60 min and 90 min respectively, which are comparable with that of *Lp group but not as effective as that of *Lp.
In Eddy’s hot plate, though $Lp (15.41 %) and UL (14.62 %) showed some increase in MPR %, but it was negligible considering their respective basal normal values. NPLs, PLp and BPLs showed very low or negligible rise of MPR % both in Eddy’s hot plate and tail immersion test.
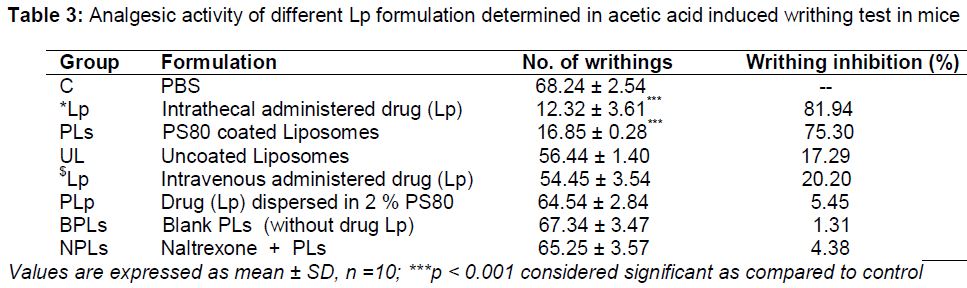
In acetic acid induced writhing study (), *Lp showed significant decrease (p < 0.001) in the number of writhing and the protective effect was 81.94 %. The PS80 coated liposomal formulation (PLs) was also found to reduce the no. of writhing significantly (p < 0.001) and the protective effect was 75.30 %. Uncoated liposomes (UL) showed writhing inhibition of about 13.96 %. Blank PLs (BPLs) and $Lp showed very low inhibition. The effect of naltrexone was prominent in this writhing inhibition study which showed very negligible percentage of writhing inhibition (4.38 %) and this would be attributed to its antagonistic property against the drug Lp in the brain, delivered by PLs.
Dose response study
Dose response study of PLs () gave a sigmoidal curve. The dose of 25 µg/kg Lp produced max MPR (58.42 %). With further increase of dose to 30, 35 and 40 µg/kg, a plateau was observed, indicating no further increase in MPR. This sigmoidal curve shows that the minimum effective dose of Lp in PLs formulation is 25 µg/kg for maximum analgesic activity.
Discussion
Our results have shown the potential analgesic effect of PLs in both Eddy’s hot plate and tail immersion tests. This substantiates positive delivery of loaded Lp byPS80 coated liposome.
Naltrexone is an opioid antagonist which displaces the opioid agonist from receptors binding site. In both central analgesic models when naltrexone was given along PLs, it showed very low or negligible rise of MPR% and is probably due to the displacement of centrally delivered Lp from the opiod receptors. This confirms the presence of Lp receptors in the brain and any rise in pain threshold was only by central Lp action and not by peripheral mechanisms [24]. Therefore, NPLs provide substantial evidence for the presence of Lp in the mouse brain.
The acetic acid induced abdominal constriction (writhing test) has been widely used for evaluating analgesic screening [22]. Intraperitoneal administration of acetic acid showed abdominal constriction resulting in stretching of hind limbs known as writhing response [25]. Pain sensation in this writhing method is triggered by local inflammatory response resulting in the release of free arachidonic acid, prostaglandins and other mediators into the peritoneum which in turn stimulate nociceptive neurons [26]. This writhing response can be counteracted by CNS opiate receptor agonist. In this study, *Lp and PS80 coated liposomal formulation (PLs) was found to reduce the number of writhing significantly.
In some studies, it has been reported that coating of nanoparticles with PS80, which adsorbs apolipoproteins, enables the liposomes to penetrate the BBB [27]. Thus, the analgesic effect of PLs could be due to the presence of PS80.However, further research is needed to confirm this finding. But still further research is needed to find reasons behind the analgesic effects of PLs.
Conclusion
The findings of this study show that PS 80 coating has a fair potential of inhibiting P-gp efflux for delivery of Lp to brain and exhibits central and peripheral analgesic activity. The lipid-based delivery (PLs) with PS80 coat would help endocytic uptake transcytosis across BBB. This novel approach can further be explored for many potential drugs which face BBB restrictions.
Declarations
Acknowledgement
References
Archives


News Updates